At 13 years and 4 months the Chemistry Development Kit (CDK) is reasonably mature for a software project. Over the years there have been many changes in development practices as the code base evolved. This post is a departure for the usual algorithms and performance tests and looks at a recent and major change in the CDK development process.
On Monday, Egon, Nina and I made the final alterations that changed the build system from Ant to Maven. This change has been in the works for a long time and has been suggested multiple times. The actually migration has taken about a years worth of planning.
This system was customised and specific to the CDK which, in my opinion, made it a significant barrier to contributions. I know that personally I struggled to understand what was going on at compile time. A highly customised build process makes it not only difficult for a human to comprehend but also any automated tooling (Integrated Development Environments, IDEs). Superficial support has been provided for Eclipse and Netbeans editors but neither correctly interrupted the modules and relationships between them.
There is still more work to do on the module organisation, for example, CMLWritier is the the 'libiocml' module whilst the CMLReader is the 'io' module. The modules are mainly organised by their dependancies but in future it may be beneficial to organise by function. Normally classes with similar dependencies have similar function but this isn't always the case. An example of this is seen with the LINGOFingerprinter and SignatureFingerprinter in the 'smiles' and 'signatures' modules rather than the 'fingerprint' module.
On Monday, Egon, Nina and I made the final alterations that changed the build system from Ant to Maven. This change has been in the works for a long time and has been suggested multiple times. The actually migration has taken about a years worth of planning.
If you want to have a play with the new build system yourself I've put together a brief guide that also describes how to import the project into several popular IDEs - Building CDK. The project README also summarises the command line usage.
I download CDK releases and use it my project, what has changed?
If you are using the CDK as a dependancy you should not notice any difference. The library and bundled dependencies will still be distributed at each release. If you are also using maven then CDK module artefacts have been deployed for last few releases. These are by far the easiest way to use the library as dependency versioning is managed maven and newer releases can be automatically downloaded. Please see the project README for repository details.I build the CDK source and use it my project, what has changed?
The source code is now built with maven - the README summarises the steps. As with releases, SNAPSHOT artefacts will be deployed to a remote repository (currently EBI).
I have modifications to the CDK that I apply, what has changed?
If your patches are Git commits then these can still be applied. Git will sort out and use the correct file locations to any modified files. If the patch creates new files these will need to be moved manually to the correct location.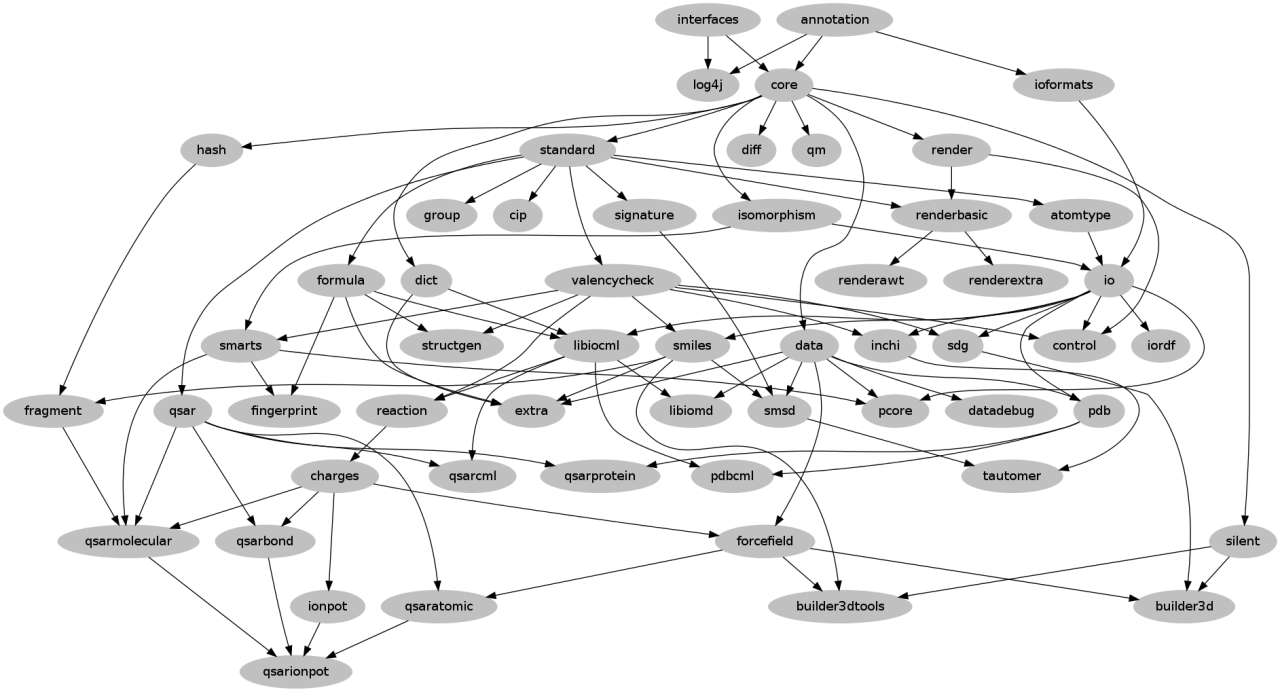 |
| CDK Modules in Dec 2013 - Egon W, Bits of Blah |
Existing project structure
Prior to Monday the project code was organised under a single root folder. The Ant build would then read instructions in the source code and assemble the modules during compilation. This approach allowed progressive partition the code into modules over an extended period. Without this system we would not have be able to convert to maven at all.This system was customised and specific to the CDK which, in my opinion, made it a significant barrier to contributions. I know that personally I struggled to understand what was going on at compile time. A highly customised build process makes it not only difficult for a human to comprehend but also any automated tooling (Integrated Development Environments, IDEs). Superficial support has been provided for Eclipse and Netbeans editors but neither correctly interrupted the modules and relationships between them.
Separate source trees
The most noticeable difference in the project is each module now has a separate source tree. This allows easier reasoning about the contents of module and provides a visual cue about the modular structure. Below we can see the 'cip' (Cahn-Ingold-Prelog) module source tree only contains the classes relevant to the module. Separate source trees are not specific to maven and we could still use Ant. The main benefit is that Maven supports and encourages this kind of structuring by default. |
| Source code in the CIP module |
Super modules
The Maven build also allows us to define groupings of the existing modules. These intermediate modules group the code base in a few digestible sections. You can see these groupings at the root level in the repository - https://github.com/cdk/cdk/. There are five groups and an additional misc/ module for the left overs. I'm planning to write a more in depth guide for the wiki but here is a quick overview of what is present in each.
- base/ - API and implementations of domain objects and central algorithms to handling chemical information
- descriptor/ - fingerprinters, qsars and signatures for describing and characterising attributes of a compound.
- storage/ - reading and writing of chemical compounds from multiple file formats
- tool/ - structure diagram generation, smarts, smsd, hashcodes, tautomer and tools that either answer a question directly, manipulate input or compute intrinsic properties
- display/ - rendering of 2D depictions


cc1.png)

co.png)

C1=CC=CC2=C1NC=N2.png)
C1=CC=CC2=C1N=CN2.png)
C(O)=O.png)
C(O)=O.png)



